Constipation is an important problem in the
pediatric emergency department for many reasons. It is one of the most common
pediatric complaints, accounting for 3% of primary care visits. There are many
causes for constipation (Table 13.1), some rare and some very common (Table
13.2). Occasionally, the presentation of constipation is atypical, with chief
complaints that superficially seem unrelated to the gastrointestinal tract
(Table 13.3). Although relatively rare, some causes of constipation are
potentially life threatening and need to be recognized promptly by the
emergency physician (Table 13.4). In addition, constipation may produce
symptoms that mimic other serious illnesses such as appendicitis.

Although constipation most commonly is defined
as decreased stool frequency, there is not one simple definition. The stooling
pattern of children changes based on age, diet, and other factors. Average
stooling frequency in infants is approximately 4 stools per day during the first
week of life, decreasing to 1.7 stools per day by 2 years of age, and
approaching the adult frequency of 1.2 stools per day by 4 years of age.
Nevertheless, normal infants can range from 7 stools per day to 1 stool per
week. Older children can defecate every 2 to 3 days and be normal.
It is easier to define constipation as a problem
with defecation. This may encompass infrequent stooling, passage of large and/or
hard stools associated with pain, incomplete evacuation of rectal contents,
involuntary soiling (encopresis), or inability to pass stool at all.
PHYSIOLOGY
The passage of food from mouth to anus is a
complex process. The intestine relies on input from intrinsic nerves, extrinsic
nerves, and hormones to function properly. Normal defecation involves voluntary
and involuntary components. Disruption of any of these can result in
constipation. The colon is specialized to transport fecal material and balance
water and electrolytes contained in the feces. When all is functioning well,
the fecal bolus arrives in the rectum formed but soft enough for easy passage
through the anus. Normal defecation requires the coordination of the autonomic
and somatic nervous systems and normal anatomy of the anorectal region. The
internal anal sphincter is a smooth muscle, which is innervated by the
autonomic nervous system. It is tonically contracted at baseline. It relaxes in
response to the arrival of a fecal bolus in the rectum, allowing stool to descend
to the portion of the anus innervated by somatic nerves. At this point, the
external anal sphincter, striated muscle under voluntary control, tightens
until the appropriate time for fecal passage. Before defecation, squatting
straightens the angle between the rectum and the anal canal, allowing easier
passage. Voluntary relaxation of the external anal sphincter allows passage of
the feces, and increasing intraabdominal pressure via Valsalva aids the
process.
EVALUATION AND DECISION
The evaluation of the child presumed to have
constipation should begin with a thorough history and physical examination.
Special attention should be paid to the age of the patient, duration of
symptoms, timing of first meconium passage after birth, changes in frequency and
consistency of stool, stool incontinence, pain with defecation, rectal
bleeding, presence of abdominal distention and/or palpable feces, and a rectal
exam to assess anal position, sphincter tone, widening of the rectal vault, and
presence of hard stool. A complaint of constipation is not sufficient for
diagnosis. A decrease in stool frequency or the appearance of straining is
often interpreted as constipation. The physician should be aware of the
grunting baby syndrome, or infant dyschezia, in which an infant grunts, turns
red, strains, and may cry while passing a soft stool. This is the result of
poor coordination between Valsalva and relaxation of the voluntary sphincter
muscles. Examination reveals the absence of palpable stool in the rectum or
abdomen. Complaints of constipation not supported by history or physical
examination are called pseudoconstipation (Fig. 13.1).
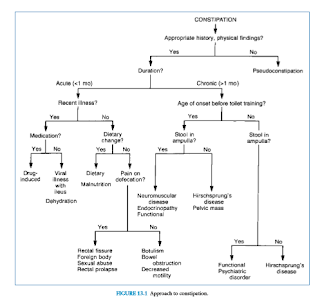
Acute Constipation
Constipation is not a disease; it is a symptom
of a problem. Constipation is acute when it has occurred for less than 1
month’s duration. The patient’s age and the duration of the constipation are
important when determining the cause and significance of the problem.
The infant younger than 1 year of age with true
constipation is particularly concerning. Potential causes include serious
diseases such as dehydration, malnutrition, and infant botulism. A recent viral
illness accompanied by dehydration from excessive water loss through vomiting,
diarrhea, fever, and increased respiratory rate can precipitate acute
constipation in an infant. Adynamic ileus or decreased intake after
gastroenteritis may cause slower transit time through the colon, which can also
lead to hard stools. Anal fissures and/or diaper rash after a bout of diarrhea
may precipitate painful defecation, resulting in stool retention. In this case,
the infant may assume a retentive posture consisting of extension of the body
with contraction of the gluteal and anal muscles.
Excessive intake of cow’s milk, inadequate fluid
intake, and malnutrition should all be uncovered by a complete dietary history.
Recent courses of medication cannot be overlooked because many can cause
constipation (Table 13.5). Ingestion of lead is also a potential and serious
reason for constipation.
Infantile botulism commonly presents with acute
constipation, weak cry, poor feeding, and decreasing muscle tone. Acute constipation can also be a symptom of a bowel obstruction, but is
normally a less prominent feature than other symptoms.
Acute constipation in the child older than 1
year of age occurs for many of the same reasons as in the infant. History may
reveal recent viral illness or use of medication, as well as the presence of
underlying illness, such as neuromuscular disease. Physical examination suffices
to rule out anal malformations and other physical problems that could result in
trouble defecating.
Chronic Constipation
Constipation of more than 1 month’s duration in
an infant, although probably a functional problem, is especially concerning and
should prompt consideration of an underlying illness. Spinal muscular atrophy,
amyotonia, congenital absence of abdominal muscles, dystonic states, and spinal
dysraphism, which cause problems with defecation, can be readily diagnosed with
history and physical examination.
Anorectal anomalies occur in approximately 1 in
2,500 live births. Anal stenosis causes the passage of ribbonlike stools with
intense effort. Diagnosis is made by anal examination, which demonstrates a
tight, constricted canal. The condition is treated by repeated anal dilations,
sometimes over several months. The anus can be covered by a flap of skin,
leaving only a portion open for passage of stool. This “covered anus” may
require anoplasty with dilation. Anterior displacement of the anus is believed
to cause constipation by creating a pouch at the posterior portion of the
distal rectum that catches the stool and allows only overflow to be expelled
after great straining. The treatment may be medical or surgical.
Hirschsprung’s disease, or congenital
intestinal aganglionosis, is rare but must be considered in the constipated
infant because it has the potential to cause life-threatening complications.
The incidence is 1 in 5,000 live births, with a male:female predominance of
4:1. As a result of failure of migration of ganglion cell precursors along the
gastrointestinal tract, there is the absence of ganglion cells in the
submucosal and myenteric plexuses of the affected segment. The absence of
ganglion cells leaves the affected segment tonically contracted, blocking
passage of stool. The segment proximal to the blockage dilates as the buildup
of stool progresses. In most cases, the child never feels the urge to defecate
because the blockage is proximal to the internal sphincter and anal canal.
In Hirschsprung’s disease, abdominal
examination often yields a suprapubic mass of stool that may extend throughout
the abdomen. Rectal examination reveals a constricted anal canal with the
absence of stool in the rectal vault, commonly followed by expulsion of stool
when the finger is removed. The combination of palpable abdominal feces and an
empty rectal vault is abnormal and must be further investigated.
Megacolon in Hirschsprung’s disease can lead to
enterocolitis characterized by abdominal distension; explosive stools, which
are sometimes bloody; and fever progressing to sepsis and hypovolemic shock.
Enterocolitis represents a major cause of mortality in this condition.
Of infants with Hirschsprung’s disease, 80% are
diagnosed within the first year of life. A history of late passage of meconium
is often found (Table 13.6). However, if the involved segment is relatively
short, the diagnosis may be delayed. If suspected, diagnosis is supported by
unprepped barium enema, which typically demonstrates narrow bowel rapidly
expanding to a dilated area. This transition zone represents the location where
the aganglionic, tonically contracted bowel meets the dilated, innervated
bowel. In disease where only a short segment of bowel is involved, barium enema
may miss the transition zone and anal manometry aids in diagnosis. Confirmation
is achieved by demonstration of aganglionosis on biopsy.

Hypothyroidism in the infant may present with
constipation. Water-losing disorders such as diabetes insipidus and renal
tubular acidosis may also contribute to this condition. Cystic fibrosis can
present with constipation alone; when there is a history of delayed passage of
meconium and Hirschsprung’s disease has been ruled out, evaluation by a sweat
test is indicated.
Chronic constipation in the older child is
overwhelmingly likely to be functional constipation. Typically, a cycle of
stoolwithholding starts when the child disregards the signal to defecate and
strikes a retentive posture—rising on the toes and stiffening the legs and
buttocks. This maneuver forces the stool out of the anal canal and back into
the rectum, which subjects the fecal bolus to further absorption of water. The
longer the stool sits, the more likely defecation is to be painful and
traumatic. This reinforces stool-withholding behavior, creating larger and
harder stool in the rectum.
Over time, in functional constipation, the
rectum dilates and sensation diminishes. Eventually, the child loses the urge
to defecate altogether. Watery stool from higher in the gastrointestinal tract
can leak around the large fecal mass, causing involuntary soiling, or
encopresis. This may be misconstrued as diarrhea or as regression in the
toilet-trained child. Many parents consult a physician at this point. Other
reasons parents seek medical attention for their children are abdominal pain,
anorexia, vomiting, and irritability.
Peak times for constipation to develop are when
routines change. Toilet training represents a major alteration in the toddler’s
routine. It is also a time when the child and caregiver battle for control.
Another problematic time is after starting school, when a child may be
uncomfortable using an unfamiliar bathroom or unable to adapt to a lack of
privacy. Involvement with friends or games may distract a child from the signal
to defecate. Painful defecation from streptococcal perianal disease or sexual
abuse must be remembered as potential precipitants of stool withholding. In
addition, functional constipation can be associated with dysfunctional urinary
voiding and urinary tract infections.
A history supportive of functional constipation
includes retentive posturing, infrequent passage of very large stools, and
involuntary soiling during the peak ages. Physical examination typically
reveals palpable stool in the abdomen. The back should be inspected for skin
changes over the sacral area, which would suggest spinal dysraphism. Normal
deep tendon reflexes and strength in the lower extremities in conjunction with a
normal anal-wink reflex virtually excludes neurologic impairment. The anus
should be normal in placement and appearance. Rectal examination typically
yields a dilated vault filled with stool. Abdominal flat-plate x-ray can be
helpful but is not necessary (Fig. 13.2). Failure to thrive is not associated
with functional constipation and, if present, should prompt further
investigation.
Although functional constipation encompasses
most cases of chronic constipation in the child older than 1 year of age, the
less common causes must always be considered.
As in the infant, endocrine abnormalities and
other disorders can cause and present as constipation. Hypothyroidism is often
associated with constipation, as well as with sluggishness, somnolence,
hypothermia, weight gain, and peripheral edema. Diabetes mellitus produces
increased urinary water loss and, in the long term, intestinal dysmotility,
which can lead to constipation. Hyperparathyroidism and hypervitaminosis D,
which lead to increased serum calcium, cause constipation through decreased
peristalsis. Celiac disease is also recognized as a cause of chronic
constipation.
Rarely, an abdominal or pelvic mass may present
with chronic constipation. Careful abdominal examination will demonstrate the
mass. Rectal masses may present similarly. Follow-up again is emphasized
because a mass that does not resolve after clearance of impaction needs further
evaluation. Hydrometrocolpos can present with constipation and urinary
frequency; therefore, a genital examination is indicated in girls to document a
perforated hymen. One must also remember that intrauterine pregnancy is a
common cause of pelvic mass and constipation in adolescent girls.
Children with neuromuscular disorders often
develop chronic constipation. Myasthenia gravis, the muscular dystrophies, and
other dystonic states can predispose children to constipation through a number
of mechanisms. A detailed history and physical examination should recognize
most neuromuscular problems, allowing symptomatic treatment to be provided.
Psychiatric problems must not be forgotten in
the evaluation of constipation. Depression can be associated with constipation
secondary to decreased intake, irregular diet, and decreased activity. Many
psychotropic drugs can cause constipation. Anorexia nervosa may present with
constipation because of decreased intake or metabolic abnormalities, and
laxative abuse can cause paradoxical constipation.
TREATMENT
Simple acute constipation in an infant should
be treated initially with dietary changes (Table 13.7). Decreasing consumption
of cow’s milk, possible formula change, and increasing fluid intake when
appropriate may be enough to alleviate the symptoms. In addition, supplementing
the diet with sorbitol as found in prune, pear, white grape, and apple juice
can be helpful to soften the stool and improve stool passage. If dietary
measures are not sufficient, lactulose or barley malt soup extract (Maltsupex®)
may be useful as osmotic agents. Historically, Karo® corn syrup had been used
as an osmotic agent, but its use has fallen out of favor after concerns that
the syrup may contain spores of Clostridium botulinum. Stool lubricants such as
mineral oil should not be used in children younger than 3 years of age and
should also be avoided in some older children when aspiration is a risk.
Polyethylene glycol solutions such as MiraLax® have gained increased use in the
outpatient setting (see discussion below). When perianal irritation or anal
fissures are present, local perianal care may decrease the risk of painful
defecation, which, in turn, may decrease stool-retentive behavior. Follow-up is
the most important aspect of treating simple constipation.
Therapy for acute functional constipation in
the child older than 1 year of age should be the same as that for the infant,
with dietary changes and stool softeners as mainstays; however, attention
should also be paid to psychological factors such as recent stress that may be
complicating the situation.
Treatment for chronic constipation in the
infant younger than 1 year of age should include ongoing dietary measures
including several daily servings of pureed fruits and vegetables, sorbitol-containing
juices, and possible formula change. If dietary measures alone are insufficient
to control symptoms, a daily stool softener such as lactulose can be used to
help maintain soft stool passage and a glycerin suppository can be used on
occasion to disimpact the rectum, although this should not be used regularly.
Although safety data are still emerging, polyethylene glycol (PEG) 3350
(MiraLax®, GlycoLax®) may also be a safe and effective treatment for chronic
constipation in infants. Loening-Baucke and colleagues studied 20 children and
Michail and colleagues studied 12 children younger than 1 year of age, who were
safely and successfully treated with PEG 3350 used for several months or more.
Although more safety data is needed to make specific recommendations, this will
likely become one of the therapeutic options for this age range.
Treatment (Table 13.7) for chronic functional
constipation in the child older than 1 year of age begins with disimpaction and
evacuation of the stool remaining in the colon. This is accomplished with
either oral or rectal therapy or a combination of the two. A study by Youssef
et al. demonstrated that in fecally impacted children whose palpable stool mass
did not extend above the level of the umbilicus, PEG 3350 at a dose of 1 to 1.5 g per kg per day (up to
a maximum of 100 g per day) given for 3 days was an effective method of
disimpaction and evacuation. Other oral options include lactulose, sorbitol,
senna, bisacodyl, PEG electrolyte solution, magnesium hydroxide, and magnesium
citrate. Rectal disimpaction can be accomplished with hypertonic phosphate
(Fleet®) enemas or bisocodyl suppositories. A mineral oil enema administered
the night before the first phosphate enema may soften existing stool, allowing
less painful passage. Phosphate enemas are typically dosed at one adult-sized
enema (133 mL) for patients 3 years and older, and one pediatric-sized enema
(66 mL) for those 1 to 3 years of age. Phosphate enemas should not be used in
children younger than 1 year. The enema may be repeated, spaced 24 hours apart,
with a maximum of three total doses. Subsequent doses should only be given if
evacuation of the previous dose has occurred. Phosphate enemas should be used
with caution in patients with dehydration, prolonged enema retention, or renal
impairment because such use has rarely been associated with severe
hyperphosphatemia, hypocalcemia, and tetany, and consequent life-threatening
complications. Tap water and soapsuds enemas should be avoided because of the
possibility of water intoxication. Enemas will disimpact but oral agents are
often needed in addition to produce full bowel evacuation. If there is no
response after 2 days, more aggressive disimpaction under physician supervision
is indicated. Oral phospho-soda preparations
should never be used in children and have been removed from the market
secondary to serious electrolyte abnormalities.
The long-term maintenance phase of therapy,
which is equally as important as the disimpaction and evacuation phase,
involves nonstimulant osmotic laxatives, lubricants, fluids, fiber, and
behavioral therapy. Laxatives include hyperosmolar agents such as lactulose and
PEG 3350. Lubricants such as mineral oil and Kondremul® are helpful to
lubricate the intestine for easier passage of stool. These should only be used
in children older than 3 years of age and those without a high risk for
aspiration. Some have advocated the use of fatsoluble vitamin supplementation
when mineral oil products are used, but there is little evidence to suggest
this is truly necessary. Increasing fluid and fiber intake is also critical to
longterm success in treating constipation. Table 13.8 outlines the recommended
daily fiber intake for different ages. Fiber should be increased gradually
toward the goal to minimize side effects of flatulence. Regular toileting should
be encouraged with positive reinforcement in the school-age child. Toilet
training should be discontinued in the training toddler until retentive
behaviors have improved. Education of patients and parents about the
pathophysiology of constipation, the etiology of encopresis when present, and
the expectations of therapy are vital. Close follow-up is a mainstay of
treatment. Successful therapy may take months to years to complete.
Approach to the Patient
with Severe Chronic Constipation
Disimpaction and evacuation of stool in the
patient with severe chronic constipation or one who has failed simple therapy
presents a challenge, particularly in the emergency department setting. A
series of phosphate enemas may not be sufficient to disimpact a larger stool
mass. Use of PEG with electrolytes solution (GoLYTELY®) as a lavage either
orally or via nasogastric tube at a dose of 10 to 25 mL per kg per hour up to
1000 mL per hour until stool is clear may be helpful to treat more severe
impactions. This method should be done in the hospital under supervision of a
physician with close monitoring of the patient’s volume and cardiovascular
status and electrolytes. Risks may be higher in patients with complex medical conditions
such as cardiac disease. Gastrograffin or N-acetylcysteine enemas may be an
additional method of disimpaction, especially in the case of distal intestinal
obstructive syndrome as occurs in patients with cystic fibrosis. In cases of
very severe fecal impaction, surgical disimpaction may be necessary. The use of
milk and molasses enemas in children is falling out of favor in many
institutions as a result of safety concerns following several case reports of
serious adverse events, including one death, after administration. The other components
of constipation therapy apply as already outlined previously and in Table 13.7.


Suggested Readings
Benninga MA, Voskuijl WP, Taminiau JAJM.
Childhood constipation: Is there new light in the tunnel? J Pediatr Gastroenterol
Nutr 2004;39:448–464.
Constipation Guideline Committee of the North
American Society for Pediatric Gastroenterology, Hepatology and Nutrition.
Evaluation and treatment of constipation in infants and children:
recommendations of the North American Society for Pediatric Gastroenterology,
Hepatology and Nutrition. J Pediatr Gastroenterol Nutr 2006;43:e1–e13.
Loening-Baucke V. Prevalence, symptoms and
outcome of constipation in infants and toddlers. J Pediatr 2005;146:359–363.
Loening-Baucke V, Krishna R, Pashankar DS.
Polyethylene glycol 3350 without electrolytes for the treatment of functional
constipation in infants and toddlers. J Pediatr Gastroenterol Nutr
2004;39:536–539.
Michail S, Gendy E, Preud’Homme D, et al.
Polyethylene glycol for constipation in children younger than eighteen months
old. J Pediatr Gastroenterol Nutr 2004;39:197–199.
Nurko S, Youssef NN, Sabri M, et al. PEG3350 in
the treatment of childhood constipation: a multicenter, double-blinded,
placebo-controlled trial. J Pediatr 2008;153:254–261.
Pashankar D, Loening-Baucke V, Bishop W. Safety
of polyethylene glycol 3350 for the treatment of chronic constipation in
children. Arch Pediatr Adolesc Med 2003;157:661–664.
Walker M, Warner BW, Brilli RJ, Jacobs BR.
Cardiopulmonary compromise associated with milk and molasses enema use in
children. J Pediatr Gastroenterol Nutr 2003;36:144–148.
Youssef N, Peters JM, Henderson W, et al. Dose
response of PEG 3350 for the treatment of childhood fecal impaction. J Pediatr
2002;141(3):410–414.














